
If you work in battery electrolyte R&D, chances are you have seen results in the lab, or even in computer simulations, that beg for explanations that are not forthcoming from available data.
If you could understand the origin of an electrolyte formulation’s high or low rate capability, electrochemical stability, ionic conductivity or electrode wettability, you could continue your search for better electrolytes in a more rational way, guided by insight rather than blind search or gut feeling.
Similar questions were plaguing me when I was working as a PhD student in Patrik Johansson’s research group at Chalmers University of Technology, in the context of highly concentrated electrolytes. These had been observed to possess several highly unusual properties that made them interesting for industrial application: unusually wide electrochemical stability, both on the reductive and oxidative side and regardless of whether EC was present, as well as high rate capabilities and low volatility. These properties were at the time poorly understood.
I was tasked with gaining a deeper understanding of the class of highly concentrated electrolytes by computational modeling. I tried to make progress developing simplified theoretical models of the real dynamics, but was stumped by the almost complete lack of scientific knowledge about these electrolytes’ structure and solvation dynamics. Theoretical modeling builds from assumptions, but it was completely unclear what assumptions were called for.
To overcome this hurdle I instead turned to molecular dynamics (MD), specifically ab initio MD (AIMD) in order to not rely on force fields of questionable applicability. Instead of making a priori assumptions, I developed trajectory analysis methods for discovering the emergent structure and solvation dynamics of simulated electrolytes.
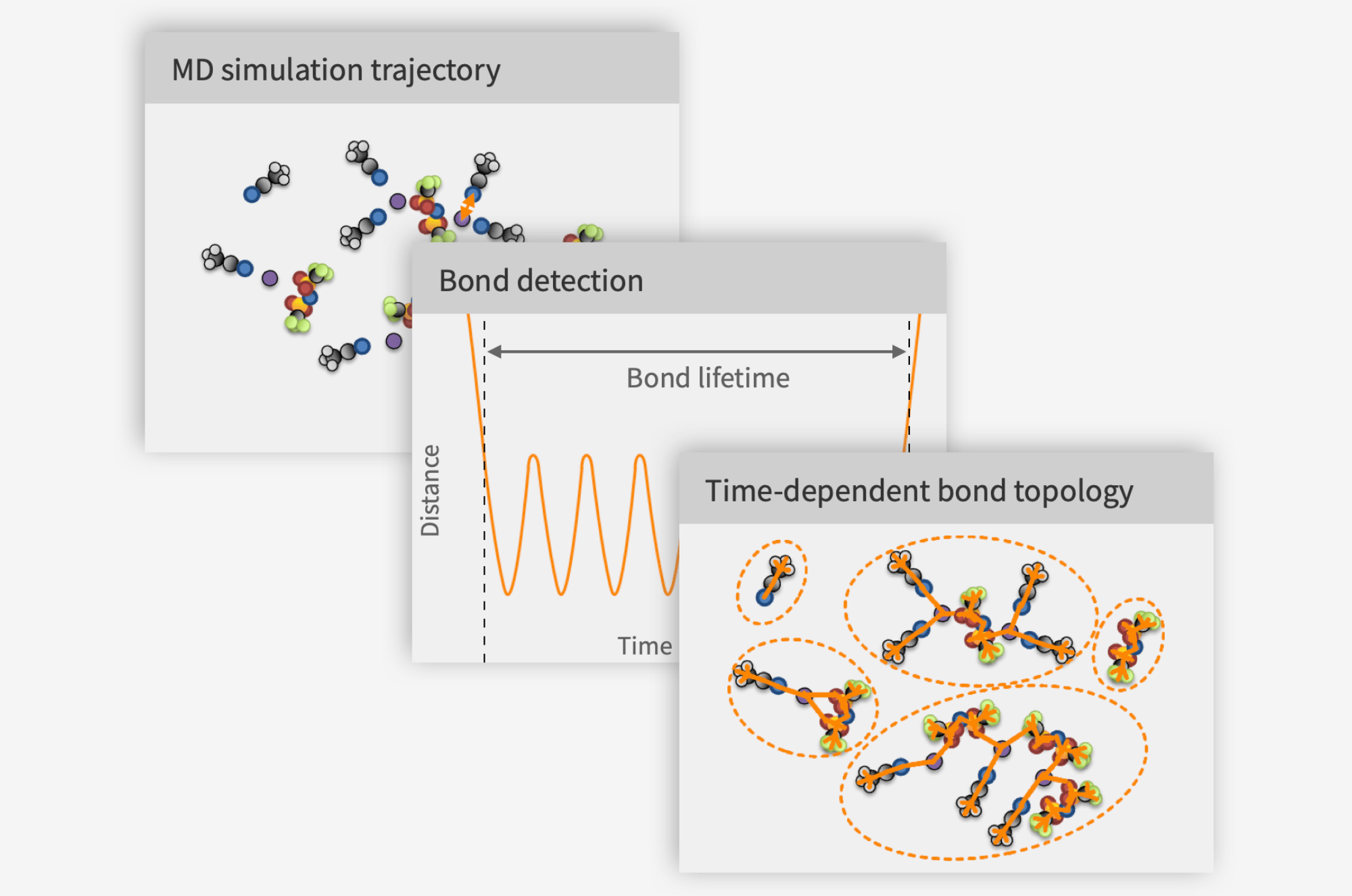
The key insight was that if I could find what moves together in an MD trajectory and how that changes over time, then I could detect what I would otherwise have to assume. After a few failed attempts, a working solution took shape:
Find windows of time wherein pairs of atoms are closer than a fraction of the sum of their van der Waals radii.
Within each such window, compute the average distance between the atoms. If this is close enough to the first peak in the partial radial distribution function (pRDF) of the pair, there may be a bond.
It is possible that two atoms fulfill the previous two criteria not because they are bound directly, but because they are both bound to a common third atom. To exclude such spurious bonds, make sure the line between the atoms does not pass through a cone defined by a given angle distended around a shorter bond involving any of the atoms in the pair.
This method, which has since been granted as an SE patent (SE 2051245-5) was the kernel around which Compular formed. Our full approach to trajectory analysis is shown schematically below:
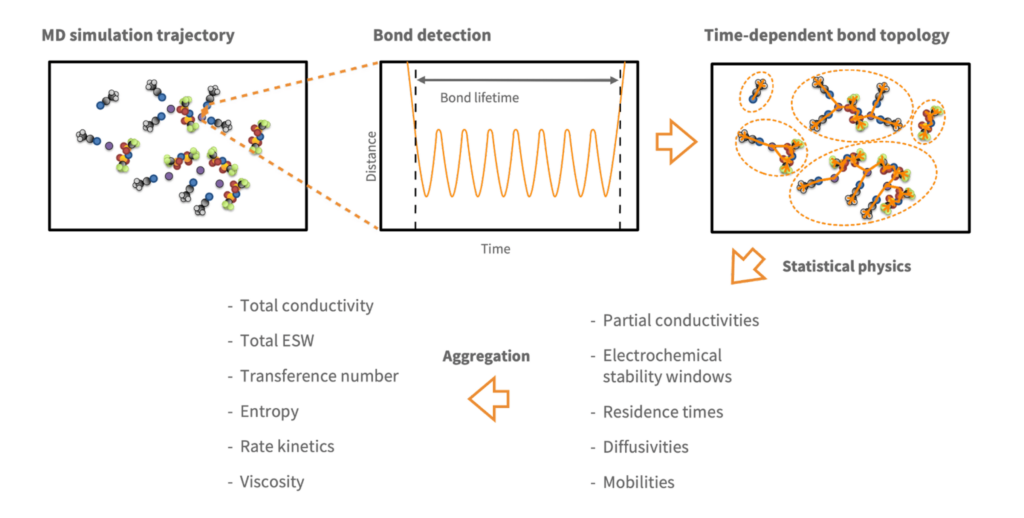
Based on how the bond detection algorithm works, bonds are discovered no matter their nature: covalent, ionic or anything else. Both intramolecular and intermolecular bonds can therefore be detected. By extending the software with graph functionality, we further enabled it to recognize molecules and to capture the speciation by tracking which distinct solvation shells, ion pairs and aggregates form. The final ingredient of our trajectory analysis approach is to characterize each dynamic species by statistical physics to compute its diffusivity, partial conductivity, mean lifetime, electrochemical stability window, etc. By aggregating these properties across species, overall system properties like total conductivity and viscosity can be computed.
If you would like to hear more about Compular’s solutions and how they can help you in your R&D work, feel free to contact us at to set up a meeting.
If you would like to read more about our technology, take a look at the list of references linked here.



